Fig. 1
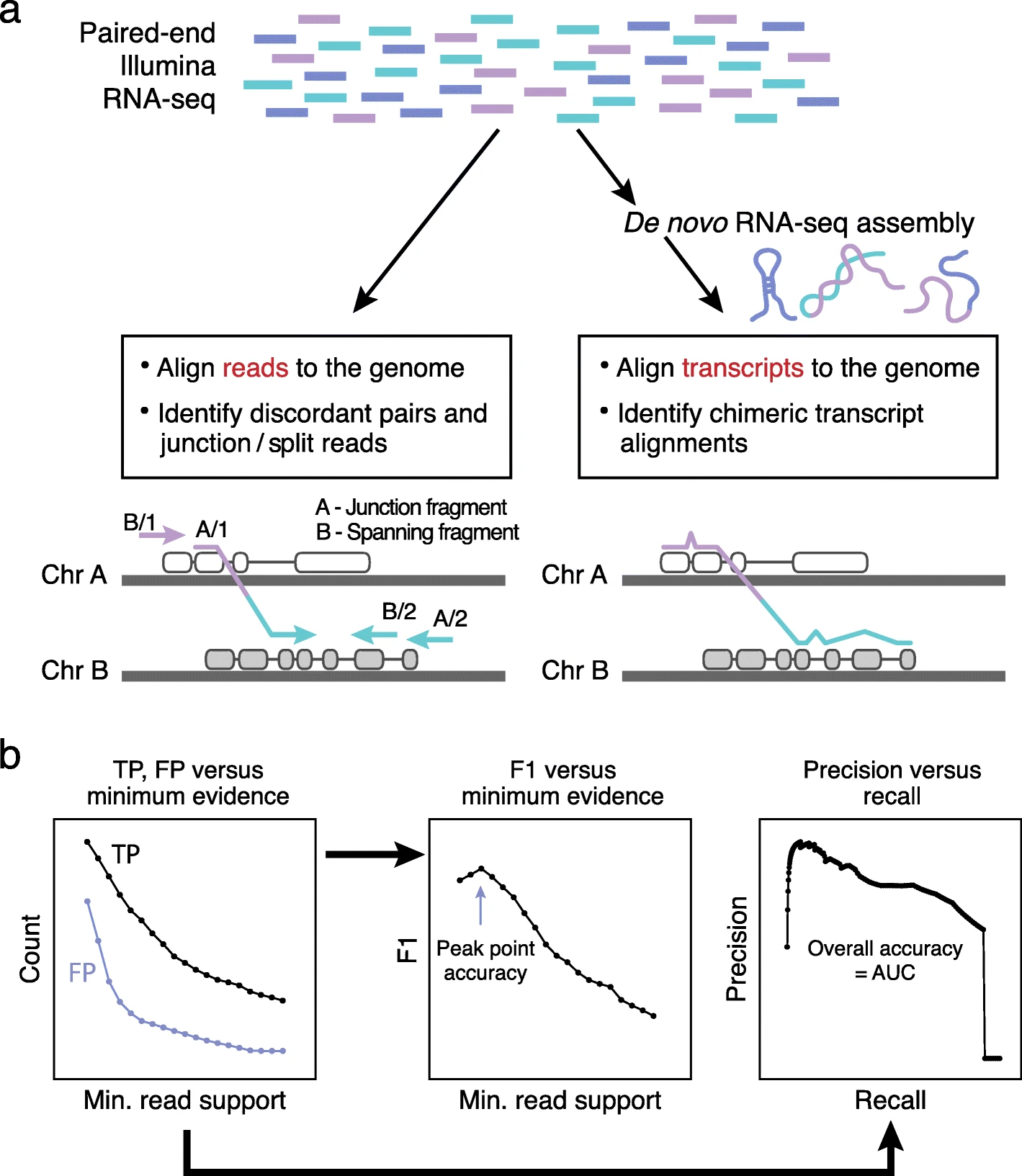
Methods for fusion transcript prediction and accuracy evaluation. a The two general paradigms for fusion transcript identification include (left) mapping reads to the genome and capturing discordant read pairs and chimeric read alignments and (right) performing genome-free de novo transcript assembly followed by identification of chimeric transcript alignments. b Given a well-defined truth set of fusions, true- and false-positive predictions are tallied according to minimum threshold for fusion-supporting reads. F1 accuracy values are computed at each minimum evidence threshold to determine the threshold that yields peak prediction accuracy for each method. Similarly, precision and recall values are computed at each minimum evidence threshold, plotted as a precision-recall curve, and the area under the curve (AUC) is computed as a measure of overall prediction accuracy